

Paper 3: General and Practical Techniques in Chemistry
This is a culmination of information that may be useful for the paper 3 in new specification (2015) A Level Edexcel chemistry, which will be a test of practical techniques and understanding - such as knowledge of what increases accuracy or reliability of experiments; how to construct equipment (such as reflux and distillation); calculating uncertainties... all using knowledge from topics 1-19. I used information from Units 6 and 3 of the IAL (practical papers) to inform this. (This was created before the first year of the spec so we didn't have much material to work on)
EDIT ** sorry if some of the image links are now broken as this is quite an old resource which I am not updating any more, most can be found by similar Google search**
- Created by: LouiseG
- Created on: 09-05-17 13:50
Safety
Working safely is about identifying risks, and planning how to reduce them. Chemicals in chemistry will come labelled with pictograms, which inform you of the hazard posed by that substance. 
For example, ethanol is flammable. An appropriate precaution would be to keep it away from naked flames (Bunsen burners). So, in esterification, you may heat your carboxylic acid and alcohol with a heating mantle, or using a bunsen burner but with a water bath. Concentrated ammonia solution should be handled in a fume cupboard, wearing gloves. The gas is toxic and the solution corrosive. Note that concentration does matter- for example, an acid may be corrosive at high concentrations but an irritant when dilute. Sometimes you may have to evaluate why a "hazardous substance" in fact does not pose much of a risk in an experiment, such as a flammable liquid. Answers are often to do with the fact only very small (or dilute) volumes are used, so the risk of a large fire or explosion is low.
Core technique #1 : Reflux
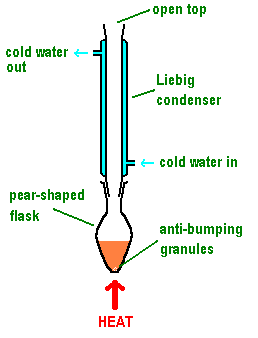
Reflux is used for the prolonged heating of volatiles that would otherwise escape the reaction vessel as gases. An upright condenser is fitted over a round or pear-shaped flask, which is heated by a bunsen burner (often with a water bath) or heating mantle. Important notes include: water goes in the bottom and out the top of the condenser (so it is forced through); anti-bumping granules should be used to ensure smooth boiling; the apparatus should be sealed around the condenser and flask but NOT at the top, otherwise gases could build up to a dangerous pressure. Your products should not escape out the top as they should condense before reaching it.
Core technique #2: Distillation (Simple)
This is a technique used to separate compounds with large differences in boiling points (ideally over 25oC). It is the quickest form of distillation. The setup is similar to reflux but with a sidearm condenser. The top is now sealed (otherwise the gases would escape) but the opening on the receiving adaptor prevents pressure build up. Ideally, a pure product should be collected at just +- 1oC of its data book boiling point. Note again that water is forced through the condenser against gravity. The thermometer's bulb should be opposite the mouth of the condenser to measure the temperature of the gas just distilling over. Sometimes in place of the thermometer, there may be a vessel (a "dropping funnel") which delivers the reactant slowly into the round-bottomed flask throughout the reaction (distillation with addition)

Core technique #3: Steam distillation
This is used to separate an insoluble liquid from an aqueous solution. Steam is passed into the vessel of a setup similar to distillation to agitate both layers, bringing more particles to the surface of the mixture so they evaporate more easily, below their boiling point. This fact means it can be good for distilling substances that decompose on heating, as the risk of this is reduced at lower temperatures. The product distills with water so it will have to be purified further with a drying agent (e.g. MgSO4, CaO).
Core technique #4: Fractional distillation
This is used to separate more substances with more similar boiling points. The continual evaporation-condensation up the column means that the compounds will separate out better. Notice again this is sealed at the top, with a thermometer to measure the temperature of the substance distilling over. This is the slowest form of distillation, but the best at separating.
Core technique #5: Solvent extraction
If you want to remove a particular substance from a mixture, you can use solvent extraction. A solvent which is immiscible with the solvent containing the desired product is added in a separating funnel and mixed, opening the tap at regular intervals. The solvent is chosen so that the desired product is far more soluble in it than the original one. This is left to settle into two layers. Next, you need to consider densities. The less dense substance will be the top layer. Organics tend to be less dense than aqueous solutions. Consider which layer you want, then use the tap to drain into two separate flasks. Now, the product will have to be separated again from the solvent, this time by fractional distillation. Note that the yield of product is higher if the solvent is added in small batches rather than all at once. 
Core technique #7: Drying
Drying refers to removing water, which is often an impurity in synthesis. For an organic solid, it may be enough to simply leave it in a warm place, and then patted dry on filter paper. For organic liquids, the water can be removed with a drying agent. These are substances that will absorb the water but not react with the organic liquid, and are usually anhydrous metal salts, such as MgSO4 and Na2SO4. They look powdery when anhydrous, but become crystalline upon reaction with water. The drying agent is added until it remains powdery (suggesting it can absorb no more water - all removed). The liquid will turn from cloudy to clear. This can be done in a desiccator for an organic solid. This is a sealed container containing the wet solid, with the drying agent. As the drying agent is a solid, it is easily removed from liquid products by decantation (pouring the product off) or filtration.
Filtering under reduced pressure
This is a technique which filters a solid product by using an idea of a vacuum, created by the rapid flow of water, which 'pulls' the liquid off the solid and filters it much faster. The product is placed on filter paper over a Buchner funnel, which has small holes in the funnel to draw down the liquid. This is fitted tightly over the Buchner flask which collects the filtrate.


Testing for purity
Once your product has been obtained, then you will want to test its purity to check you have synthesised the right compound! For a solid, the melting temperature can be measured; for a liquid the boiling temperature. For a liquid, distillation is used to find the boiling point.
For a solid: A capillary tube (small tube of glass) is sealed at one end by placing the end in a bunsen burner flame. The open end is then pushed into a small amount of the solid. The tube is inverted and tapped so that the solid falls to the bottom. This is then taped to a thermometer and placed in an assembly containing a liquid with a higher boiling point than the substance being tested. It should also be ideally non-flammable and colourless so the melting can be seen easily. The liquid is slowly heated until the solid begins to melt. The range over which it melts is recorded. A sharp melting point over 1-2 degrees, in agreement with the data book value, suggests a pure compound. A melting range that begins lower than the databook value, and over several degrees, indicates impurities present.
Core technique #9: Titration
Carrying out a titration properly is vital for a chemist. This came up in Year 1, but here are a summary of the key titration points:
- Fill the burette at eye-level (mentioned before). The burette should also be washed through with the substance that will be in it, e.g. NaOH solution if NaOH will be in the burette. If water is used, this will dilute the substance, change its concentration and the titration won't be accurate.
- Read off the bottom of the meniscus (The curved surface of the liquid) at eye level when recording titres. This is how the apparatus is calibrated. For a burette, if you read off the top of the meniscus for both your first and last readings, it technically wouldn't matter (the difference between the values would cancel), but, for a pipette, where you take one reading, a failure to fill up to the bottom of the meniscus along the graduation line would mean the measured volume would be incorrect. Potassium manganate (VII) is a commonly used in redox titrations, but because it is a very dark purple, it can be difficult so see the graduation marks on the burette. Specially made burettes, therefore, have white markings instead.
Chemical Analysis: Displacing Halides
More reactive halogens (towards the top of the group) will displace less reactive halides from solution, for example
KI(aq) + 1/2 Cl2 (g) ---> KCl (aq) + 1/2 I2 (g)
The halogens are more soluble in organic compounds such as cyclohexane than water, so if cyclohexane is added (which will form a layer on top of the (aq) solution) then the halogen that had been displaced will dissolve in this layer, as a distinctive colour -
Chlorine is pale green (displaced by F-)
Bromine is orange (displaced by Cl- , F-)
Iodine is violet (displaced by F- , Cl- , Br-)

Chemical Analysis: Ignition
Igniting an organic compound could be one way of identifying what kind of compound it is (though it is very inconclusive).
A smoky flame indicates an unsaturated compound as there is a high carbon to hydrogen ratio. Examples would be alkenes, but this is especially used for compounds containing a benzene ring.
A clean flame indicates a short chained, saturated compound. These include shorter alkanes.
No residue left over suggests that the compound has a low Mr
This should be done in a fume cupboard.
Equipment you must know
There are certain pieces of equipment you should know how to use from your experience of your core practical work. Some you will know already from previous studies, such as beakers, conical flasks and volumetric flasks, however organic synthesis apparatus (such as condensers, still heads, round-bottomed flasks) may be new.
![]() Conical flasks are used for containing liquids and are particularly useful in titrations as they are shaped so that the reagents should fall back into the reaction vessel. They are not used for measuring out volumes.
Conical flasks are used for containing liquids and are particularly useful in titrations as they are shaped so that the reagents should fall back into the reaction vessel. They are not used for measuring out volumes.
Beakers are multi-purpose but are often used for dissolving solids before addition to a conical flask. They can also be used to prepare water baths.
Equipment
 Measuring cylinders are used to measure out volumes, usually to a lower degree of accuracy than a pipette or burette.
Measuring cylinders are used to measure out volumes, usually to a lower degree of accuracy than a pipette or burette.
 Volumetric flasks are used to measure out a set (one only) volume accurately. They have a thin neck with a graduation line. The solution should be filled up until the bottom of the meniscus rests on the line.
Volumetric flasks are used to measure out a set (one only) volume accurately. They have a thin neck with a graduation line. The solution should be filled up until the bottom of the meniscus rests on the line.
Equipment
 A funnel is used to channel a liquid into a container and can be used with filter paper to filter the liquid. Some funnels do not have stems, so the product cannot crystallise in the stem and block it.
A funnel is used to channel a liquid into a container and can be used with filter paper to filter the liquid. Some funnels do not have stems, so the product cannot crystallise in the stem and block it.
 Buchner funnel and flasks are used to filter out a solid under reduced pressure. The flask creates a vacuum which sucks the liquid through the filter paper. The side tubing goes to a running tap, where the running water creates a suction to generate the reduced pressure. This is faster and removes more solvent than filtration under gravity (above)
Buchner funnel and flasks are used to filter out a solid under reduced pressure. The flask creates a vacuum which sucks the liquid through the filter paper. The side tubing goes to a running tap, where the running water creates a suction to generate the reduced pressure. This is faster and removes more solvent than filtration under gravity (above)
Equipment
![]() Test tubes are used to contain reagents. They can be easily bunged. Boiling tubes are similar but can be heated over a bunsen burner also.
Test tubes are used to contain reagents. They can be easily bunged. Boiling tubes are similar but can be heated over a bunsen burner also.
 A drop pipette is used to transfer very small volumes of fluids that do not need to be measured very accurately, e.g. to add a few drops of indicator in a titration, or to just fill a volumetric flask to the graduation line (the last few drops just as the liquid enters the thin neck of the flask).
A drop pipette is used to transfer very small volumes of fluids that do not need to be measured very accurately, e.g. to add a few drops of indicator in a titration, or to just fill a volumetric flask to the graduation line (the last few drops just as the liquid enters the thin neck of the flask).
Equipment

A graduated pipette is used to transfer accurate set volumes of liquids by having a marked graduation line on the thin stem of the pipette. The liquid is drawn up the pipette using a pipette filler.
![]() A burette is used to measure volumes of solutions added by having a scale, often from 0 - 50cm3. (with 0 at the top) Burette measurements are taken to 2 d.p, with the last digit a 0 or 5.
A burette is used to measure volumes of solutions added by having a scale, often from 0 - 50cm3. (with 0 at the top) Burette measurements are taken to 2 d.p, with the last digit a 0 or 5.
Equipment
 A separating funnel is used to separate immiscible solutions (that have formed layers). It has a tap at the bottom that can be opened to run off a layer. The bung on the top can be removed to relieve pressure if a gas is evolved.
A separating funnel is used to separate immiscible solutions (that have formed layers). It has a tap at the bottom that can be opened to run off a layer. The bung on the top can be removed to relieve pressure if a gas is evolved.

 Pear-shaped and round bottomed flasks can often be used interchangeably. They are used for heating reagents (e.g. in reflux) and collecting products (e.g. distillation).
Pear-shaped and round bottomed flasks can often be used interchangeably. They are used for heating reagents (e.g. in reflux) and collecting products (e.g. distillation).
Equipment
![]()
A condenser is used to cool a gas back to a liquid. It can be fitted upright over a reaction vessel when a volatile is being heated under reflux (continual heating and condensation,) or near-vertical as a side-arm condenser when a product is being condensed and collected in distillation. Water is pumped into the bottom and out through the top of the condenser. Ensure the bottom of the condenser is fully in your reaction vessel or still head so the gases don't escape.
 A still head is used to connect a side-arm condenser to the reaction vessel in distillation. The top may be bunged, with a thermometer.
A still head is used to connect a side-arm condenser to the reaction vessel in distillation. The top may be bunged, with a thermometer.
Equipment
 A receiving head/adaptor is used to connect the end of the condenser to the receiving flask (where the distillate ends up) in distillation. It has an open end to relieve pressure
A receiving head/adaptor is used to connect the end of the condenser to the receiving flask (where the distillate ends up) in distillation. It has an open end to relieve pressure
A crucible is a small, lidded ceramic pot used for heating substances, such as in thermal decomposition. They are initially heated gently to reduce the risk of the contents spitting out.
Equipment
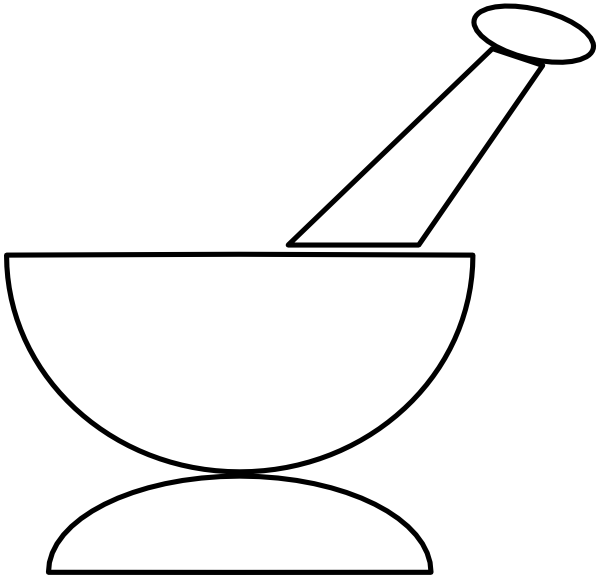 A mortar and pestle are used to grind up solids into a powder
A mortar and pestle are used to grind up solids into a powder
 Evaporating dishes are wide, shallow dishes used for drying (e.g. salts). A watch glass (slightly curved piece of glass) may also be used.
Evaporating dishes are wide, shallow dishes used for drying (e.g. salts). A watch glass (slightly curved piece of glass) may also be used.
![]() Tongs can be used to move hot beakers and test tubes
Tongs can be used to move hot beakers and test tubes
 A nichrome loop is used to transfer small volumes of solid to a flame in flame tests
A nichrome loop is used to transfer small volumes of solid to a flame in flame tests
Equipment
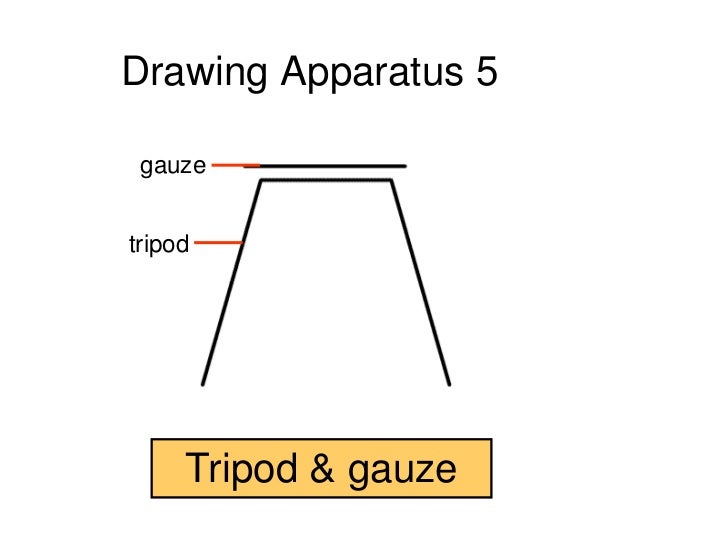 A tripod with a heat-proof gauze is used to support a beaker being heated by a bunsen burner. The tripod and Bunsen burner are placed on a heat proof mat
A tripod with a heat-proof gauze is used to support a beaker being heated by a bunsen burner. The tripod and Bunsen burner are placed on a heat proof mat
 A Bunsen burner is used as a source of heat. When the collar is "open", the flame is hotter (more oxygen) and blue, also known as roaring or non-luminous. When closed, it is a yellow "safety" flame (much more visible). Often in a diagram, you could represent a bunsen burner as an arrow labelled "heat". They should not be used (at least not without a water bath) around flammables.
A Bunsen burner is used as a source of heat. When the collar is "open", the flame is hotter (more oxygen) and blue, also known as roaring or non-luminous. When closed, it is a yellow "safety" flame (much more visible). Often in a diagram, you could represent a bunsen burner as an arrow labelled "heat". They should not be used (at least not without a water bath) around flammables.
Equipment
A fractionating column is a column packed with glass beads used in distillation. As the gases rise through the column they condense and vaporise several times on the surface of the beads, thus providing better separation.
Negative logx
This is the graph of log10(x). As you can see, the value of x cannot be negative, but the value of logx can be (see the last slide). Notice the x-intercept is (1,0), as log10(1) = 0. In fact, log(1)= 0 for all bases (i.e. not just ten). This is as anything the power of 0 is 1. 10^0 = 1.
Notice the graph is asymptotical to the y-axis. To make logx more negative, x has to get closer and closer to 0, but it won't reach it. This is as 10^x can never equal 0.
lnX
This is important for our consideration of lnX. lnX is the natural log, that is log to the base e. e is an irritational constant approximately equal to 2.718. One significance of e is that its gradient function of the graph e^x is equal to the graph of the function. This could be said in easier terms as the gradient at any point is equal to the y value for that point. (dy/dx = y )
Treat e in the same way that 10 was for log. For example, e^x = 4 would be solved by ln4 = 1.39.
e comes up in rates, for example in the Arrhenius equation:
k = Ae ^ (-Ea/RT)
Summary: Practical technique #2
2. Use a water bath, electric heater or sand bath for heating.

A water bath can be as simple as a large beaker of water, which is either heated in a kettle or over a bunsen burner on a tripod. A boiling tube can be then placed in the beaker. A heating mantle or electric heater can be used to heat a container filled with sand. It provides a more even heating for a container compared to exposing to heat from below. These methods are usually used when a direct naked flame is not favoured, or if the temperature has to be more controlled, for example, if a substance is flammable or decomposes as high temperatures.
Summary: Practical technique #3
3. Measure pH using pH charts, or pH meter, or pH probe on a data logger.

A pH probe is used to measure pHs of solutions, as in CP9 (Ka of a Weak Acid). The probe is kept in a buffer solution when not in use, and washed with deionised water and dried between measurements (so the solution does not remain on the end of the probe). It can be calibrated by finding meter readings of solutions of known pH, usually buffer solutions, and plotting a graph of meter readings against the pH (to give a calibration curve).
Remember when carrying out titration curve questions that near the equivalence point the volumes being added should be drastically smaller (e.g, only add 0.5cm3 at a time compared to 5cm3). This is as the pH will begin to change rapidly as it falls into the "steep section" of the pH curve.
Summary: Practical technique #6
6. Use acid-base indicators for weak/strong acid/base titrations.
In a weak acid-strong base titration, you want an indicator which changes colour at higher pHs, as the pH will be >7 at equivalence. To find the colour change pH, look at the pKin values. E.g., phenolphthalein (9.4). So, for a strong acid-weak base, you'd want an indicator whose pH range is at a low (acidic) pH, such as methyl orange. For a suitable indicator, the pH range has to fall in the "steep" or "vertical" section of the titration curve, when the pH changes rapidly over less than a cm3. Rember that universal indicator is not used as it has a spectrum of colours rather than a sharp colour change.
Summary: Practical technique #8
8. Use melting point apparatus
Covered on card 15. Melting point apparatus can, of course, only be used for solid products - if you have a liquid, use distillation apparatus.
Summary: Practical technique #9
9. Use thin-layer or paper chromatography
This is a technique that can be used to identify amino acids, by calculating Rf values. In paper chromatography, small spots of each sample are placed along a pencil-drawn line near the bottom of the chromatography paper. This paper is then placed in a beaker in a layer of solvent, which does not reach the pencil line. It should be perfectly parallel to your pencil line. The beaker is sealed (a "chamber" - to prevent evaporation of the solvent) and left until the solvent is a few cm from the top of the paper. It is then removed, and the final line of the solvent drawn on in paper. If the solutions were colourless (e.g. amino acids), they can be developed once the solvent is dried - such as with ninhydrin. To find the Rf value, the calculationdistance travelled by mixture/distance travelled by solvent (from the baseline - the pencil line) is used.
Thin-layer is very similar, however, the stationary phase is silica on a glass plate.
Summary: Practical technique #10
10. Setting up electrochemical cells and measure their voltage.
The key conditions for the electrochemical cells are:
- If the cell is under standard conditions, then: -all ions in solution are at 1.0moldm-3 -the temperature is 298oK -the pressure of any gas is 100kPa (1 bar)
- The salt used in the salt bridge is usually potassium nitrate. These ions are unlikely to interfere with the ions in the cell (i.e. form precipitates)
- The voltmeter should be high resistance to reduce electron flow to the external circuit (so the emf can be measured).
- If there is no metal electrode, (i.e. all are ions in solutions or gases) then a platinum electrode covered in porous platinum is used as the electrical contact (non-reactive).
- There is no ammeter or cell in the circuit. Remember, the electrochemical setup IS the cell!

Summary: Practical technique #11
11. Safely handle solids and liquids, including corrosive, irritant, flammable and toxic substances.
Covered on cards 3,4 and 5.
- Corrosive - potential to cause burns to skin and eyes. Wear gloves, goggles, lab coat. If volatile - carry out in fume cupboard
- Irritant - potential to cause inflammation to skin, eyes, throat. Wear goggles, gloves, carry out in fume cupboard or well-ventilated room
- Flammable - potential to start a fire. Don't use near a naked flame; use a water bath or electric heater instead. If it is volatile, use in a fume cupboard to remove fumes.
- Toxic - poisonous - could cause illness or death on contact, inhalation or ingestion. Wear gloves, goggles, lab coat, use in fume cupboard if volatile or a fine powder.
Put stoppers back into bottles when not in use, clear up spillages immediately, keep away from table edges etc are other common sense measures.
Comments
Report
Report
Report
Report
Report